

Jim LeNomenclatoriste
Je suis un mignon petit Traquet rubicole

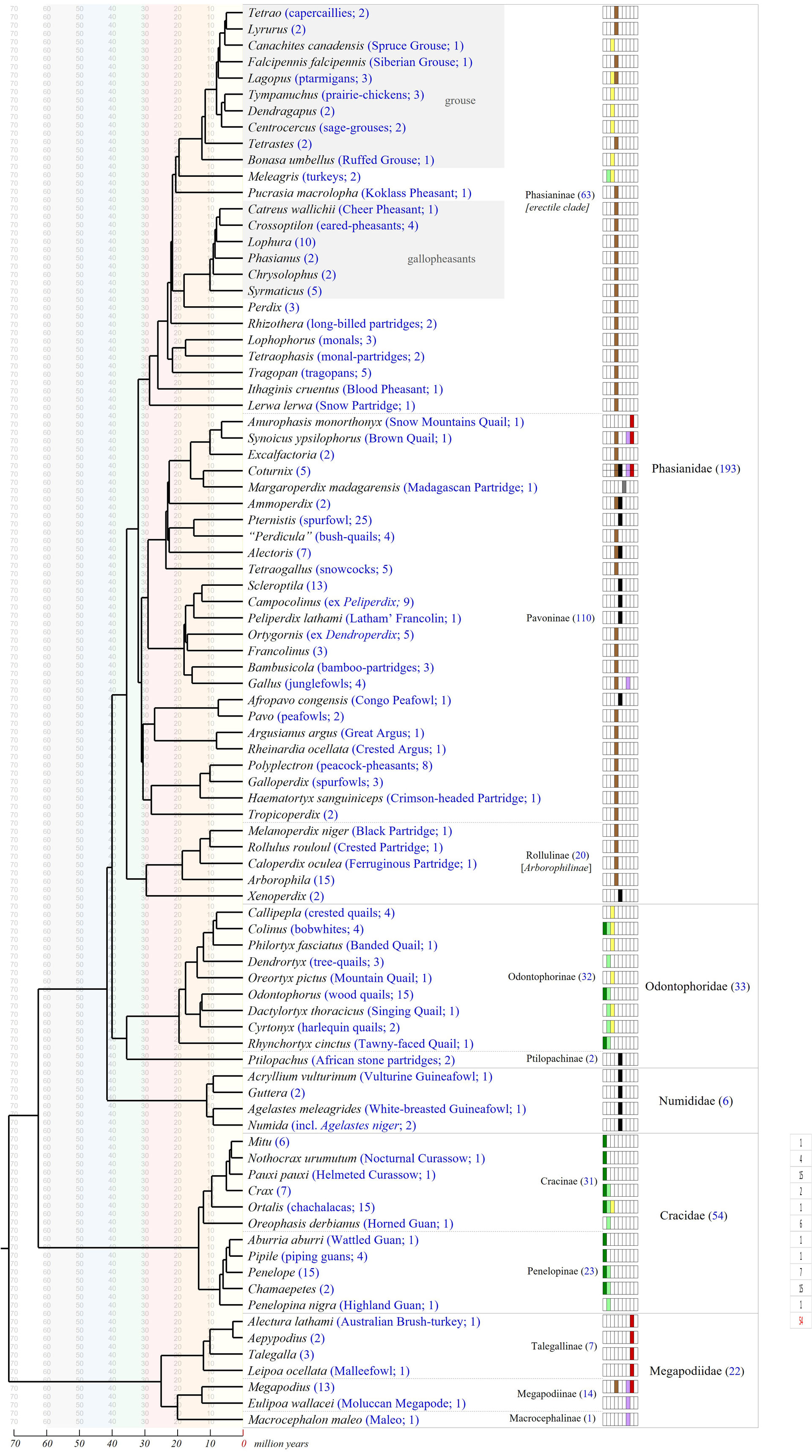
Agelastes, Pauxi and Perdicula are not monophyletic.Rebecca T. Kimball, Peter A. Hosner, Edward L. Braun. A Phylogenomic Supermatrix Of Galliformes (Landfowl) Reveals Biased Branch Lengths. Molecular Phylogenetics and Evolution, In Press, Journal Pre-proof, Available online 2 February 2021.
Abstract:
Building taxon-rich phylogenies is foundational for macroevolutionary studies. One approach to improve taxon sampling beyond individual studies is to build supermatricies of publicly available data, incorporating taxa sampled across different studies and utilizing different loci. Most existing supermatrix studies have focused on loci commonly sequenced with Sanger technology (“legacy” markers, such as mitochondrial data and small numbers of nuclear loci). However, incorporating phylogenomic studies into supermatrices allows problem nodes to be targeted and resolved with considerable amounts of data, while improving taxon sampling with legacy data. Here we estimate phylogeny from a galliform supermatrix which includes well-known model and agricultural species such as the chicken and turkey. We assembled a supermatrix comprising 4500 ultra-conserved elements (UCEs) collected as part of recent phylogenomic studies in this group and legacy mitochondrial and nuclear (intron and exon) sequences. Our resulting phylogeny included 88% of extant species and recovered well-accepted relationships with strong support. However, branch lengths, which are particularly important in down-stream macroevolutionary studies, appeared vastly skewed. Taxa represented only by rapidly evolving mitochondrial data had high proportions of missing data and exhibited long terminal branches. Conversely, taxa sampled for slowly evolving UCEs with low proportions of missing data exhibited substantially shorter terminal branches. We explored several branch length re-estimation methods with particular attention to terminal branches and conclude that re-estimation using well-sampled mitochondrial sequences may be a pragmatic approach to obtain trees suitable for macroevolutionary analysis.
Agelastes niger is sister to Numida meleagris while A. meleagrides is sister to Guttera and Acryllium. So, I have put A. niger in Phasidus as Phasidus niger
Perdicula is formed of two distinct lineage, one with P. erythrorhyncha (now Cryptoplectron erythrorhyncha), and one other with P. asiatica (include but not analyzed the following species manipurensis and argoondah which share same morphological features with asiatica).
Pauxi pauxi is close to Nothocrax ururmutum but with weak support. I didn't touch them. I don't know where placed Pauxi koepckeae.
I have placed Excalfactoria in synonymy with Synoicus, resulting this new combination Synoicus adansonii and S. chinensis.